
#1153 closed enhancement (fixed)
Handling short read transcript sequence data
| Reported by: | base | Owned by: | Nicklas Nordborg |
|---|---|---|---|
| Priority: | critical | Milestone: | BASE 3.0 |
| Component: | core | Version: | |
| Keywords: | Cc: |
Description
This may not be a "core" issue, it might be achieved with core or contributed plugins alone. As with most new technologies some core changes might be needed though.
Bob MacCallum wrote in http://www.mail-archive.com/basedb-users@lists.sourceforge.net/msg01559.html
I'm just thinking out loud about how to incorporate high throughput
transcriptome sequencing data into BASE. It's some way off, but I'm assuming
that it will be cheap and quantitative enough to replace arrays at some point
during the renewal period of our project (2009-2014).
1. Create an "array design" with all genes of interest (ideally this would be
the largest set possible, e.g. known genes + predicted genes of all
qualities, perhaps even predicted genes from the new sequence data). The
layout would be fictitious, of course (what's the minimum one can get away
with?).
2. Create a rawbioassay to correspond to each sequencing run.
Then *one* of 3a/b/c for each sequencing run/rawbioassay:
3a. Outside BASE, align the new sequences to genome or transcript sequences
and calculate "intensities" for each gene on the "array design" and dump
into a tab delimited raw data file. Attach that file to the rawbioassay
and import numeric data as usual.
3b. Upload the text file of sequences to the raw bioassay's "data file".
Create a BASE plugin to do the the alignment and quantification as in 3a,
and load the numeric data into the database.
3c. Similar to 3b, but calculate the intensities at the "create root bioassay"
step, similar to the Affymetrix RMA plugin.
4. continue with analysis as normal. biosources, samples etc can be linked to
the bioassay too, of course.
I guess a new raw data type (for "Generic" platform) would have to be
created for 3a (and 3b?) but that's not difficult.
Is it possible to go with 3a, but also attach the sequence file to the raw
bioassay (or scan?) - something like keeping tiff files for scans? Just for
documentation purposes.
Any thoughts from the community or developers?
Jari suggested starting a ticket here.
Attachments (10)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change History (50)
comment:1 by , 16 years ago
comment:2 by , 16 years ago
| Milestone: | → BASE 2.x+ |
|---|
comment:3 by , 14 years ago
| Milestone: | BASE 2.x+ → BASE 2.18 |
|---|---|
| Owner: | changed from to |
We'll try to get something going for this for the next release. We are currently working on some documents and an updated database design. We'll probably add a lot of new stuff (eg. items) to BASE. We'll look into the possibility to assign a "Project type" to a project which should make the web interface switch between different setups:
- Hide menu items that are not relevant
- Make sure annotations are inherited along the correct path
- Changes to the "item overview" function and it's validation options
- ... and probably more to come...
comment:4 by , 14 years ago
| Priority: | minor → critical |
|---|
comment:5 by , 14 years ago
| Status: | new → assigned |
|---|
comment:6 by , 14 years ago
by , 14 years ago
| Attachment: | sequencing-draft-1.txt added |
|---|
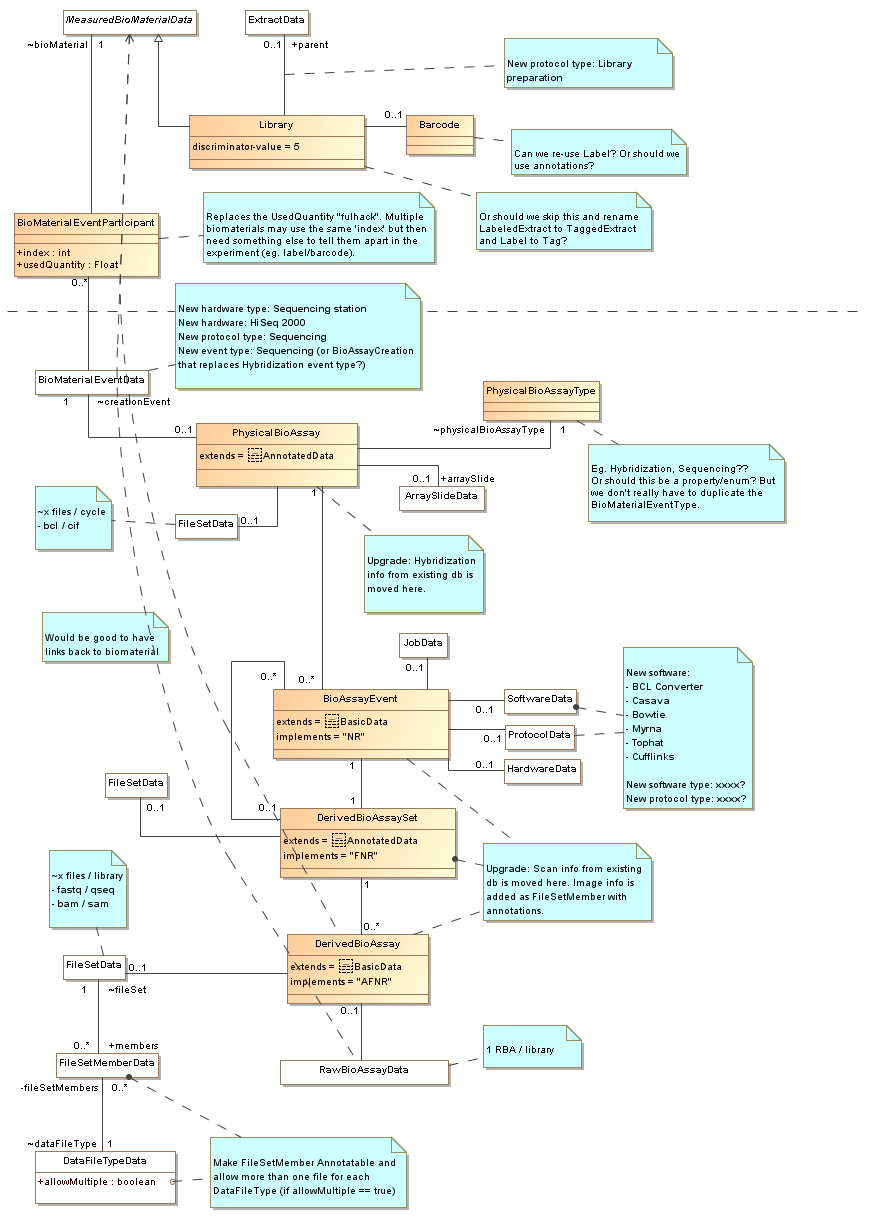
More details and thoughts about the diagram
comment:7 by , 14 years ago
by , 14 years ago
| Attachment: | sequencing-biomaterials-draft-2.png added |
|---|
Second draft: UML diagram for biomaterials
by , 14 years ago
| Attachment: | sequencing-rawdata-draft-2.png added |
|---|
Second draft: UML diagram for raw data
by , 14 years ago
| Attachment: | sequencing-draft-2.txt added |
|---|
by , 14 years ago
| Attachment: | sequencing-update-script-draft-2.txt added |
|---|
comment:8 by , 14 years ago
(In [5632]) References #1153: Handling short read transcript sequence data
Replaced Label with Tag. It re-uses much of the old code including the database table.
The code compiles and most test programs pass. Not all gui pages in the web client work. Since LabeledExtract is going to be removed functionality that is related with this may not do what is expected.
comment:9 by , 14 years ago
(In [5641]) References #1153: Handling short read transcript sequence data
Removed LabeledExtract and related classes. Extract takes it place in most situations, including a temporary link to hybridizations. All tests programs pass and the web client is more or less working. The next step is to replace Hybridization with PhysicalBioAssay.
comment:10 by , 14 years ago
comment:11 by , 14 years ago
(In [5652]) References #1153: Handling short read transcript sequence data
Replaced Scan with DerivedBioAssaySet. The code is once again in a status such that it compiles. I am working with the test programs to make sure that what has been done so far is working. The web client can't be used at the moment. The current design is a bit different from the UML. I'll update this later as I implement the remaining functionality.
comment:12 by , 14 years ago
comment:13 by , 14 years ago
(In [5657]) References #1153: Handling short read transcript sequence data
Added web pages for editing and viewing derived bioassay sets. They'll probably need to be polished up a bit.
The final link to raw bioassay is still not possible. I think we have to re-design the link somewhat. In BASE 2 we used the array index to link back to the correct (labeled) extracts. We can't use that in BASE 3 since for sequencing experiments there doesn't have to be any relation between extracts in the same lane of the flow cell. The idea was to provide a direct link to the extract, but then we loose one of the parents in 2-channel microarray experiments...
comment:14 by , 14 years ago
(In [5662]) References #1153: Handling short read transcript sequence data
Removed the old "hack" with UsedQuantity and the dummy column used to store the array index. This has now been replaced with BioMaterialEventSource which is almost a full-fledged item and it should be easier to handle and create queries using the information.
comment:15 by , 14 years ago
(In [5663]) References #1153: Handling short read transcript sequence data
Changed the way parent biomaterials are handled. The pooled property has been replaced with parentType instead. There are two reasons:
- Removing the
LabeledExtractclass has changed all toExtractitems which are not pooled. - It doesn't make sense to talk about "pooled" biomaterials when there is only a single parent.
The web gui for biosources and samples have been updated. Extracts does not work yet.
Overview loaders have been updated.
Batch importers need to be fixed. They currently assume a single parent of the parent item type.
Also added some of the button/select taglibs to provide more control over the look and feel in some cases.
comment:16 by , 14 years ago
comment:17 by , 14 years ago
comment:18 by , 14 years ago
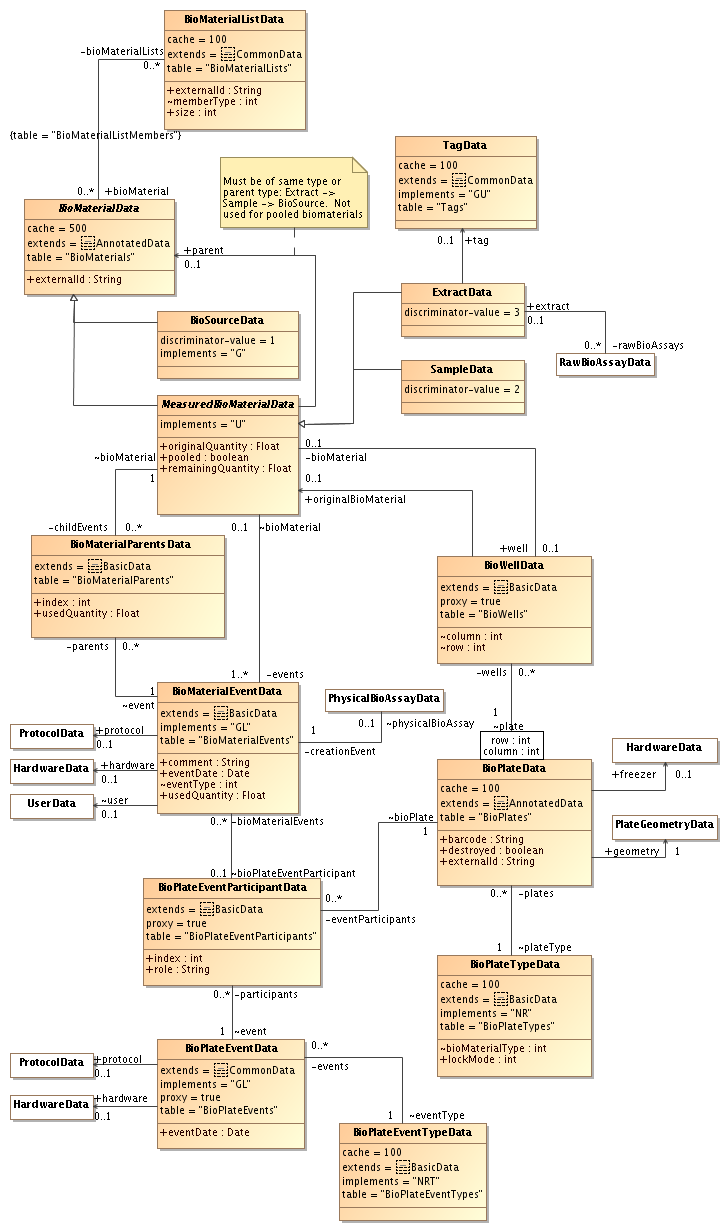
by , 14 years ago
| Attachment: | sequencing-biomaterials-3.png added |
|---|
Third version: UML diagram for biomaterials (most of this has been implemented)
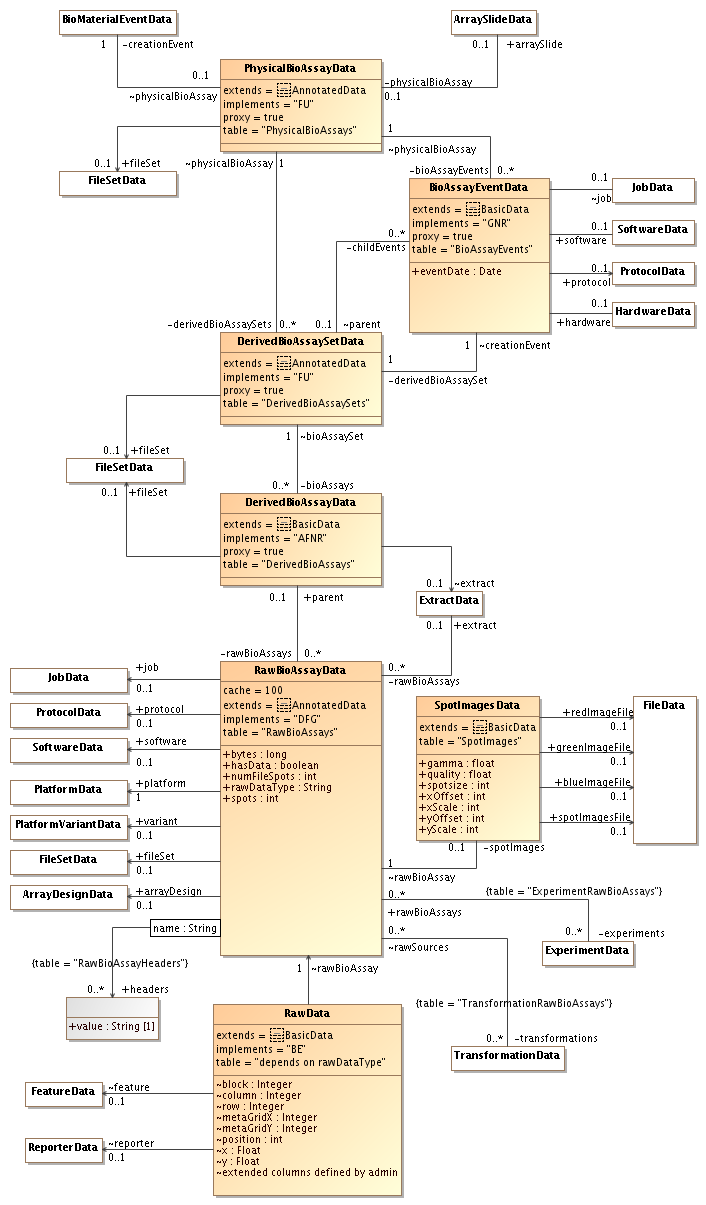
by , 14 years ago
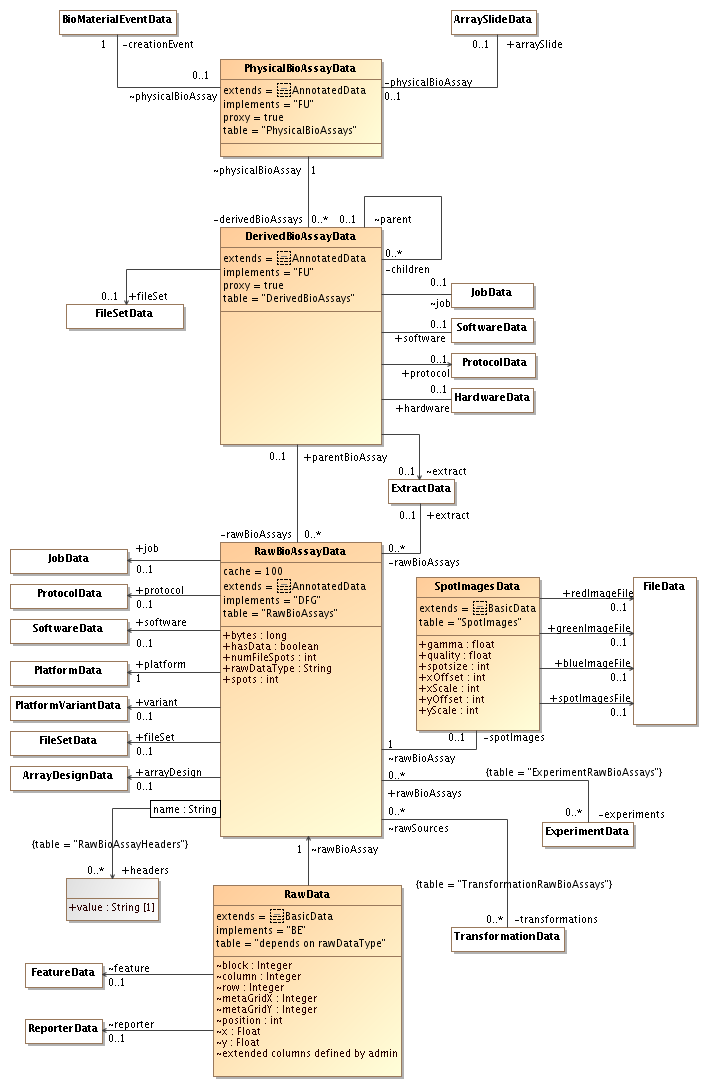
| Attachment: | sequencing-rawdata-3.png added |
|---|
Third version: UML diagram for physical bioassays+raw data (most of this has been implemented)
comment:19 by , 14 years ago
comment:20 by , 13 years ago
(In [5685]) References #1153: Handling short read transcript sequence data
Simplified the design by only keeping DerivedBioAssay between PhysicalBioAssay and RawBioAssay. This should make batch importer, validation, etc. easier to implement and also provides the better backwards compatibility with array experiments. The web gui is usable but may need improvements in some cases.
comment:21 by , 13 years ago
(In [5688]) References #1153: Handling short read transcript sequence data
Fixes some test program failures. Started to re-enable all tests in the TestItemImporter (batch importers). Also need to create a batch DerivedBioAssayImporter that replaces the ScanImporter and add support for linking to extracts from raw bioassays.
comment:22 by , 13 years ago
comment:23 by , 13 years ago
(In [5692]) References #1153: Handling short read transcript sequence data
Fixed the 'Illumina raw data importer' plug-in. As a side-effect it now has support for attaching raw bioassays to more than one scan (derived bioassay). Actually, the attachment to scans was not fully implemented in the old version and didn't work as expected.
comment:24 by , 13 years ago
comment:25 by , 13 years ago
comment:26 by , 13 years ago
comment:27 by , 13 years ago
comment:28 by , 13 years ago
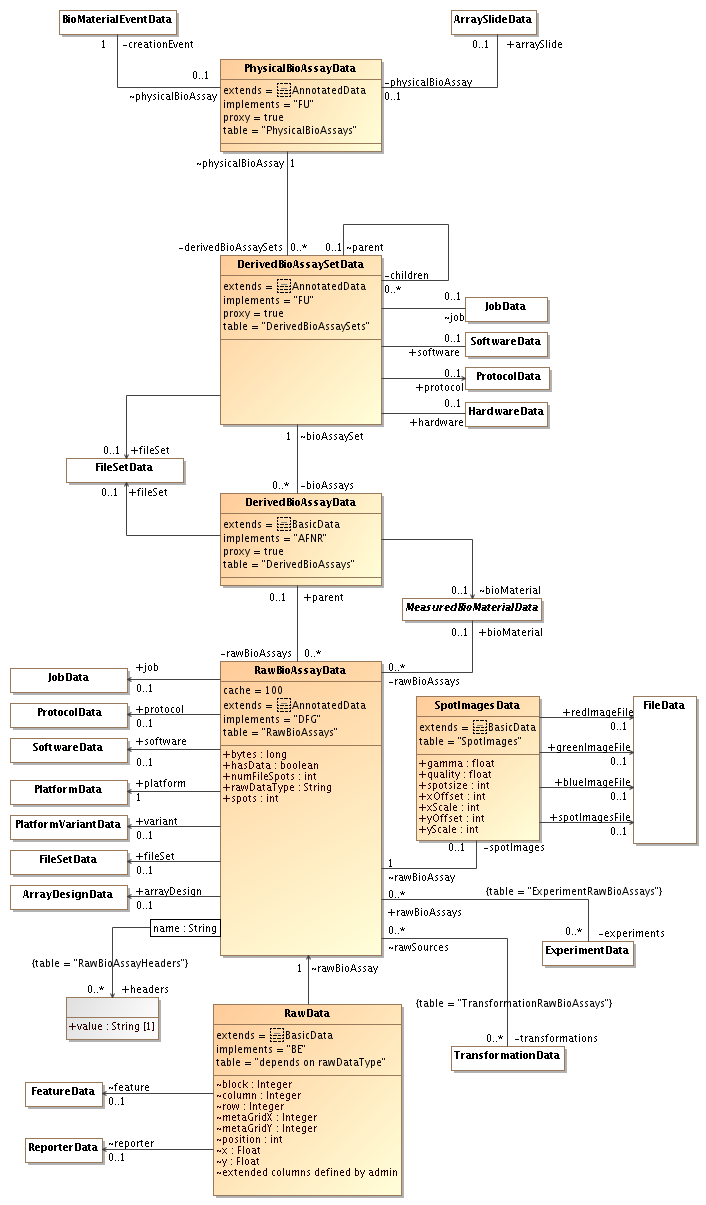
by , 13 years ago
| Attachment: | sequencing-biomaterials-4.png added |
|---|
by , 13 years ago
| Attachment: | sequencing-rawdata-4.png added |
|---|
comment:29 by , 13 years ago
The last UML diagrams simplify the path between PhysicalBioAssay and RawBioAssay. Only the DerivedBioAssay remains and it can either represent data for the entire physical bioassay (if there is no link to an Extract) or data for a single extract (if there is a link).
comment:30 by , 13 years ago
comment:31 by , 13 years ago
comment:32 by , 13 years ago
comment:33 by , 13 years ago
comment:34 by , 13 years ago
comment:35 by , 13 years ago
comment:36 by , 13 years ago
comment:37 by , 13 years ago
(In [5773]) References #1153: Handling short read transcript sequence data
- Adds a 'Cufflinks' raw data type with five columns: coverage, fpkm, fpkm_lo, fpkm_hi and status
- Define FPKM_TRACKING file type and MIME type.
- Adds left(string, index) as a JEP formula so that we can parse out the chromosome from the 'locus' column in the tracking files.
- Define two new configurations for the raw data importer that parses cufflinks isoform files.
comment:38 by , 13 years ago
(In [5807]) References #1153: Handling short read transcript sequence data
Lots of changes to item overview generation and validation. Most important are:
- Subtype validation has been improved and hopefully it now works when validating both from the top (biosource) and bottom (experiment)
- Parent extract validation of raw bioassay, derived bioassay and physical bioassay. Ensure that everything is properly linked (eg. matching extracts in all levels).
comment:39 by , 13 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
by "documentation purposes" I really mean "archival purposes"